引言
同一物种的基因组通常包含核心保守组分,以及个体或群体间存在差异的遗传物质集合——即“泛基因组(pangenome)”,简单的线性泛基因组(linear pan-genome)无法直观地描述同一物种个体基因组之间的变异,因此发展了基于图的泛基因组(graph-based pan-genome),图泛基因组以节点和路径的形式存储序列和结构变异信息,以更直观的方式存储和展示物种个体间的变异信息。应用图泛基因组进行序列比对时,相较于线性泛基因组,其比对质量在所有维度均有提升,且在变异分型过程中能检出更多结构变异(SV)以及单核苷酸多态性(SNP)和插入/缺失(Indel),并且SV长度也显著优于线性泛基因组检测结果。
基于图形泛基因组的变异鉴定简介
基于图的泛基因组可作为更广泛群体中结构变异(SV)检测和基因分型(genotyping)的新参考标准。目前绝大多数基因组分析依赖于序列比对技术,然而传统线性基因组比对工具难以适用于基于图的泛基因组比对,近年来已涌现出一批适用于图泛基因组的比对算法与软件。
vg(Variation Graph)是2018年发表在NBT上的一款基于变异和基因组构建图形泛基因组的一款主流软件,其中内置的Giraffe工具作为短读长比对工具兼具高速与精度,尤其擅长处理复杂图区域比对,是目前图泛基因组短读长比对的最佳工具,并且基于vg Giraffe比对结果,可使用vg另一个内置工具——vg call进行SV基因分型及新SV检测。
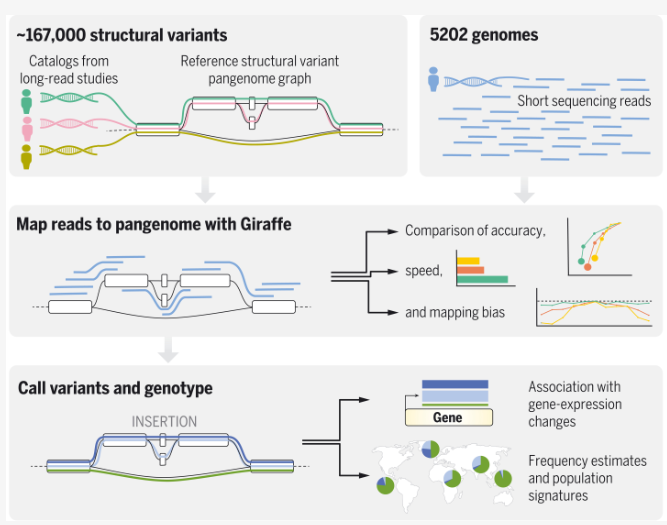
图1 基于图形泛基因组的变异检测流程图
下面将介绍基于图形泛基因组的变异鉴定的通用流程(基于Cactus方法构建的图形泛基因组):
vg安装:
用于短读长重测序数据比对的Giraffe和变异检测的pack、snarls、call均为vg的内置工具,故只需要安装vg就能完成变异检测。
conda install -c bioconda vgGiraffe:短读长重测序数据映射至图泛基因组
软件使用:
Giraffe将输入的短读长重测序数据文件*.fq和图形泛基因组文件*.gbz(基于Cactus方法构建图形泛基因组获得)进行映射,生成映射结果文件*.gam(若准备的输入文件为*.fa和*.vcf或*.gfa格式,则按照https://github.com/vgteam/vg/wiki/Mapping-short-reads-with-Giraffe 中的流程生成*.gbz文件后再使用Giraffe)。
vg giraffe -Z sample.gbz -f sample_1.fq -f sample_2.fq -t 16 > aln.gam
vg call:基于映射结果进行变异检测
软件使用:
vg pack计算reads的支持度并进行过滤生成*.pack文件;vg snarls用于分析图形泛基因组中的“Snarl”结构(Snarl 是一种拓扑结构,用于描述图形泛基因组中的嵌套变异区域),并生成*.snarl文件;最后vg call 结合*.gbz,*.pack和*.snarl文件对重测序数据进行变异检测。
# -Q 5: ignore mapping and base quality < 5vg pack -x sample.gbz -g aln.gam -Q 5 -o sample.pack -t 16# Compute the snarls(using fewer threads with `-t` can reduce memory at the cost of increased runtime)vg snarls sample.gbz > sample.snarls# Genotype the graphvg call sample.gbz -r sample.snarls -k sample.pack -a -A -t 16 -s sample_name > sample.vcf
合并变异检测结果:
软件使用:
若有多个样本的重测序数据进行变异检测,最后需要对*.vcf文件进行合并,*.vcf文件构建索引和合并需要使用bcftools软件。
#bcftools安装 conda install -c bioconda bcftools#压缩vcfbcftools view sample1.vcf -Oz -o sample1.vcf.gzbcftools view sample2.vcf -Oz -o sample2.vcf.gzbcftools view sample3.vcf -Oz -o sample3.vcf.gz# 使用 bcftools 创建索引bcftools index --threads 4 sample1.vcf.gzbcftools index --threads 4 sample2.vcf.gzbcftools index --threads 4 sample3.vcf.gz#合并bcftools merge --threads 4 -m all -O z sample1.vcf.gz sample2.vcf.gz sample3.vcf.gz -o sample.merge.vcf.gz
参考文献
-
Sirén J, Monlong J, Chang X, Novak AM, Eizenga JM, Markello C, Sibbesen JA, Hickey G, Chang PC, Carroll A, Gupta N, Gabriel S, Blackwell TW, Ratan A, Taylor KD, Rich SS, Rotter JI, Haussler D, Garrison E, Paten B. Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. Science. 2021 Dec 17;374(6574):abg8871. doi: 10.1126/science.abg8871. Epub 2021 Dec 17. PMID: 34914532; PMCID: PMC9365333.
-
Wang S, Qian YQ, Zhao RP, Chen LL, Song JM. Graph-based pan-genomes: increased opportunities in plant genomics. J Exp Bot. 2023 Jan 1;74(1):24-39. doi: 10.1093/jxb/erac412. PMID: 36255144.

关于菲沙基因
武汉菲沙基因信息有限公司(下简称:菲沙基因)成立于2012年,是一家专注于三代测序技术服务、表观遗传学技术服务、单细胞测序技术服务、基因组医学服务、生物信息学技术和软件开发应用的国家高新技术企业。公司总部位于武汉光谷生物城,在北京设立了研发中心,在广州建设了转化医学中心,又于2015年创立子公司武汉菲沙基因组医学有限公司。利用最新的三代测序技术、表观遗传学技术和单细胞测序技术,依托公司强大的生信信息平台,在人类健康、疾病防治、动植物研究、微生物应用等领域开展了研究合作,共发表项目文章500篇,累计影响因子5300+,多篇成果相继发表在《Cell》《Nature》《Science》等国际知名期刊。
菲沙基因期待与各领域的专家合作,提供快速、准确、专业的服务,共同迎接生命科学研究中的挑战。
Related Reading
相关阅读