本文最后更新于 195 天前,如有失效请评论区留言。
STEP1
主要使用到的代码分两部分,第一步脚本为shell脚本:Population_freq.sh ,可计算不同群体的等位基因频率,并生成单独文件。
需要准备的文件有两个,vcf文件以及群体分群文件
vcf_file=”186_filtered.LD.pruned.noContig.recode.vcf”
pop_file=”186sample.pop”
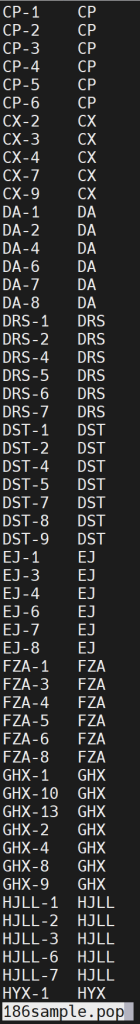
其中186sample.pop文件内容如下
脚本Population_freq.sh如下
#!/bin/bash
# 输入文件和目录
vcf_file="186_filtered.LD.pruned.noContig.recode.vcf"
pop_file="186sample.pop"
# 输出目录
output_dir="population_freq_results"
temp_dir="${output_dir}/temp_ids"
# 确保输出和临时目录存在
mkdir -p "$output_dir"
mkdir -p "$temp_dir"
# 从群体信息文件中提取独特的群体,并保存到一个临时文件
cut -f2 $pop_file | sort | uniq > "${temp_dir}/unique_groups.txt"
# 为每个独特的群体生成包含个体ID的文件
while read group; do
grep "\b$group\b" $pop_file | awk '{print $1}' > "${temp_dir}/${group}.ids"
done < "${temp_dir}/unique_groups.txt"
# 使用vcftools处理每个群体
while read group; do
(
echo "处理群体:$group"
# 使用vcftools计算等位基因频率
vcftools --vcf $vcf_file --keep "${temp_dir}/${group}.ids" --freq --out "${output_dir}/${group}_population_freq"
if [ $? -eq 0 ]; then
echo "完成群体:$group"
else
echo "计算等位基因频率失败:$group"
fi
) &
done < "${temp_dir}/unique_groups.txt"
wait
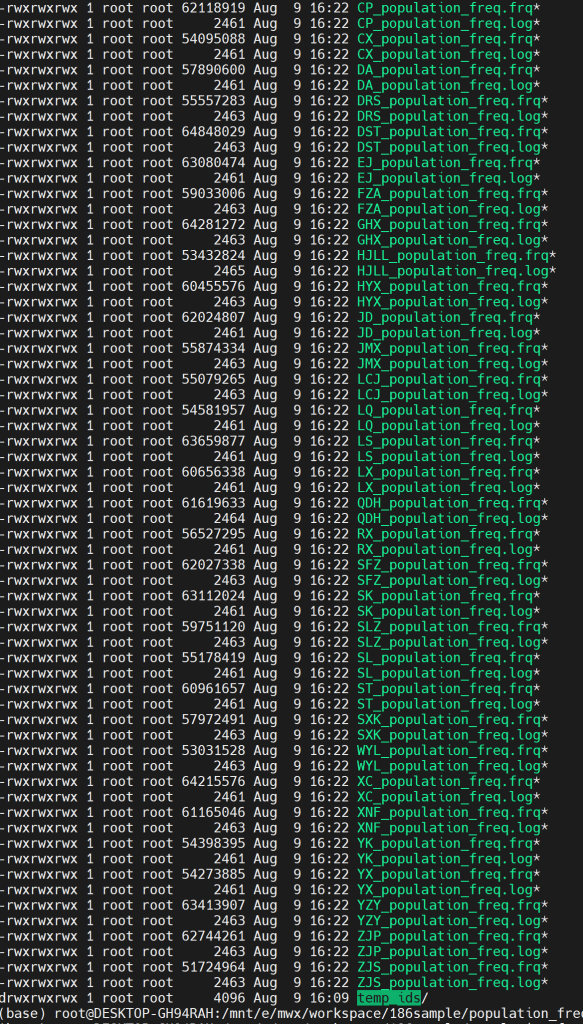
echo "所有群体处理完毕。"最终生成的文件如下
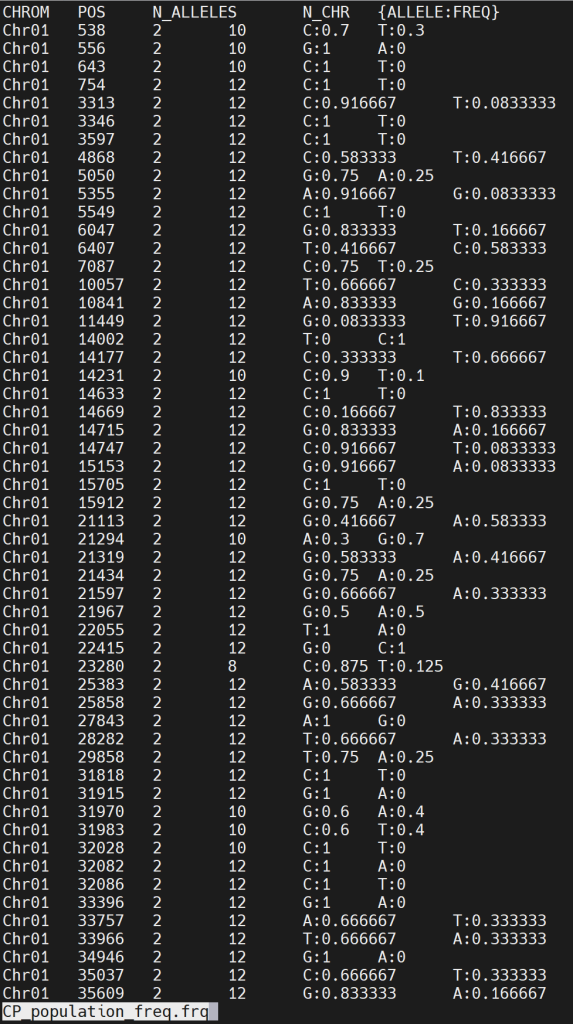
单个文件内容如下
STEP2
第二步为python脚本:combine_population_freq.py,合并不同群体的等位基因频率的单独文件,并最终保存为一个,分隔的.csv文件和一个制表符分割的.txt文件(两个文件内容一样,仅格式不同)。
需要定义的路径有两个,上一步生成的.frq文件所在的路径以及最终合并后输出的文件名
# 输入目录和输出文件
input_dir = “/mnt/e/mwx/workspace/186sample/population_freq_results”
output_file = “combined_population_freq.csv”
combine_population_freq.py 内容如下
import os
import pandas as pd
# 输入目录和输出文件
input_dir = "/mnt/e/mwx/workspace/186sample/population_freq_results"
output_csv_file = "combined_population_freq.csv"
output_txt_file = "combined_population_freq.txt"
# 获取所有 .frq 文件的列表
frq_files = [f for f in os.listdir(input_dir) if f.endswith('.frq')]
# 初始化字典来存储所有SNP信息
snp_data = {}
# 处理每个 .frq 文件
for frq_file in frq_files:
file_path = os.path.join(input_dir, frq_file)
# 从文件名获取群体名称,并去掉 "_population_freq" 部分
group_name = os.path.splitext(frq_file)[0].replace('_population_freq', '')
with open(file_path, 'r') as f:
lines = f.readlines()
# 遍历文件中的每一行数据
for line in lines[1:]: # 跳过标题行
parts = line.strip().split()
snp = f"{parts[0]}:{parts[1]}"
allele_freqs = parts[4:] # 提取所有等位基因频率对
# 提取第一个等位基因频率
first_allele, first_freq = allele_freqs[0].split(':')
if snp not in snp_data:
# 初始化FREQ列为 "allele1/allele2" 的形式
snp_data[snp] = {"FREQ": f"{first_allele}/{allele_freqs[1].split(':')[0]}"}
# 存储每个群体的第一个等位基因频率
snp_data[snp][group_name] = first_freq
# 初始化 DataFrame 并写入表头
columns = ["SNP", "FREQ"] + [os.path.splitext(f)[0].replace('_population_freq', '') for f in frq_files]
df_list = []
# 填充 DataFrame
for snp, freqs in snp_data.items():
row = {"SNP": snp, "FREQ": freqs["FREQ"]}
for group_name in columns[2:]:
row[group_name] = freqs.get(group_name, "NA")
df_list.append(row)
df = pd.DataFrame(df_list, columns=columns)
# 保存结果到 CSV 文件
df.to_csv(output_csv_file, index=False)
# 保存结果到制表符分隔的 TXT 文件
df.to_csv(output_txt_file, sep='\t', index=False)
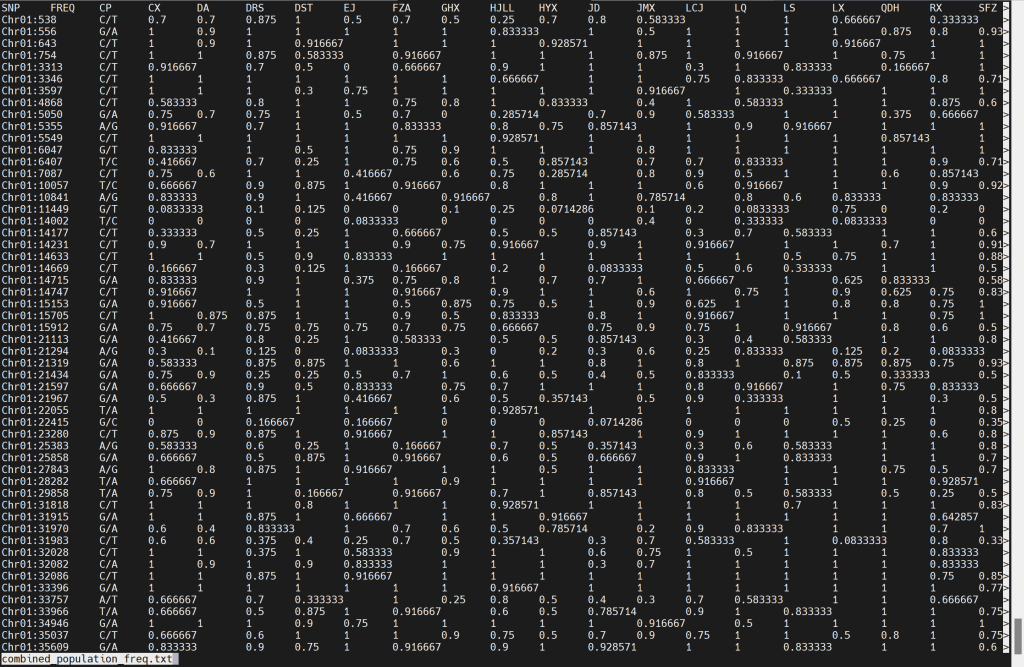
print(f"合并完成,结果保存在 {output_csv_file} 和 {output_txt_file}")最终combined_population_freq.txt文件内容如下
最终combined_population_freq.csv文件内容如下
